 Loading... Please wait...
Loading... Please wait...
- Home
- Technical Support
Technical Support
If you have Technical Support questions, please email us at info@genhunter.com, call us at 800-311-8260 x2, 615-833-0665 x2, or fill out a Contact Us form here.
Resources and Technical Notes on Differential Display
Differential Display Technical Notes
Optimal arbitrary primer length for Differential Display
The concept of differential display is to use a limited number of short arbitrary primers in combination with the anchored oligo-dT primers to systematically amplify and visualize most of the mRNA in a cell. The optimal length of arbitrary primers is determined by statistical consideration that each primer will recognize 50-100 mRNA species. To do so, these primers have to hybridize as 6-8mers (Liang and Pardee, Science. 1992, 257:967.). In practice, however, primers shorter than 9 bases failed to be used for PCR amplification (Williams et al, 1991, Nucleic Acids Res. 18:6531), probably due to the minimum contact surface between Taq polymerase with the double-stranded DNA template. Initially, arbitrary decamers (10mers) were employed for differential display in combination with two-base anchored primers. Although the method worked well in displaying the expected number of mRNA per primer combination under optimal conditions, weak signals, fewer bands, and poorer reproducibility were encountered by people who might have assembled the method under sub-optimal conditions due to the variation in the quality of RNA samples, primers, MMLV reverse transcriptase, isotopes, Taq polymerase and PCR tubes among other things. It is important to point out that the analysis of priming kinetics of arbitrary 10mers under the PCR conditions used has been extremely predictable (Liang and Pardee, 1994, Methods Mol. Gen. 5:3). These primers, as expected, hybridize initially to the first strand cDNA as 6-9mers depending on the GC content, with most of the base-pairing occurred at the 3' end of the primers.
To determine the optimal length of arbitrary primers for differential display, we have kept in mind the following essential restrictions:
- The theoretical consideration suggests that the optimal selectivity of the arbitrary primers should be provided by no more than 7 bases for each pair of primers to display 50-100 mRNAs.
- The minimum primer length for PCR is 9 bases.
- The arbitrary primers have to hybridize in a predictable way in order to rationally design a representative set of such primers to display most of mRNAs in a cell.
- The arbitrary primers in practice require 8-9 base pair matches for optimal priming.
- The redundancy (one mRNA species is represented as more than one band in each lane) should be kept to a minimum.
As a result, arbitrary primer length is increased from the sub-optimal value of 9-10 bases to 13 bases. The 13mers fulfill the above criteria for being optimal arbitrary primers (Liang et al., 1994, NAR. 22:5763). First, 13mers are perfect PCR primers as compared to longer primers in terms of being able to specifically amplify a target DNA sequence. They are much more efficient primers than 10mers, producing highly reproducible and uniform band intensities in differential display. Secondly, since the first 6-8 3' bases of these short primers provide the specificity of sequence recognition of the mRNA sequences, the arbitrary 13mers can be rationally designed in such a way that all primers are maximally different in their 7 3' base sequence (bases in bold, see table below), while the 6 5' bases are fixed and statistically can provide an additional 1.5 base pairing for each primer. So the arbitrary 13mers will hybridize as 8-9mers as required in the initial cycle of PCR amplification. Such rational primer design allows a limited number of arbitrary 13mers to be made to ensure that most of the mRNA can be represented (see figure, page 45). PCR conditions used for 10mers were preserved for 13mers to ensure the predictable nature of primer bindings. The table below shows the excellent agreement of the theory for arbitrary primer designs and the actual priming of the arbitrary 13mers in differential display based on the genes cloned.

Other companies have blindly increased the primers to 20 bases or longer in an attempt to increase the "reproducibility" of the cDNA pattern displayed. One has to remember that longer primers are used to amplify specific genes, while shorter primers are used to specifically amplify multiple genes. The longer primers under degenerate conditions will hybridize in a non-predictable way, making a rational design of a representative set of primers impossible. Although, longer primers can also generate a complex cDNA pattern which may appear reproducible, many of the bands may in fact represent the same mRNA due to the "Stickiness" of long primers when used under low stringency. In fact, A. Simpson and colleagues have used this low stringency PCR for long primers to detect sequence polymorphism of the same gene (Simpson et al., 1994, PNAS, 91:1946). To do this, they first specifically amplified a target gene of interest under high stringency conditions. Then, using the DNA sequence specifically amplified as a template and the same pair of long primers to perform PCR under low stringency (decreasing the annealing temperature), they were able to obtain a complex pattern of PCR products from a single template. Therefore the "more reproducible nature" of long primers may be misleading because of the redundancy of the PCR products amplified.
One-base versus two-base anchored primers
Dr. Peng Liang was asked how he came up with the two types of anchored primers used in differential display and which type is better. Here is his explanation:
The initial choice of using two-base anchored oligo-dT primers (Liang and Pardee, 1992, Science 257:967; Liang et al., 1993, Nucleic Acids Res. 21:3269.) instead of one-base anchored primers (Liang et al., 1994, Nucleic Acids Res.22:5763.) was due to historical rather than scientific reasons. The initial idea was to use one-base as an anchor, but the cloned murine thymidine kinase (TK) cDNA originally used as a control cDNA template had only 11 A's in its poly(A) tail. It was found that one-based anchored primer T7C up to T11C failed to amplify the TK 3' terminus in combination with an upstream primer specific to TK. Extension of one more base from the 3' end instead of the 5' end of the anchored primer was a logical choice. Interestingly, T11CA started to work successfully in PCR to amplify the expected TK cDNA template. That was how two-base anchored primers were first described.
Later, longer one-base anchored primers were shown to be much more efficient for differential display in sub-dividing the mRNA populations into three groups. One-base anchored primers have significant advantages over the two-base anchored primers in that the former reduces the redundancy of priming, eliminates the high background smearing problem for two-base anchored primers ending with the 3' "T", and reduces the number of reverse transcription reactions from 12 to 3 per RNA sample.
Theoretical estimation of the number of arbitrary primers necessary to cover most of the genes expressed in a cell
Based on the experimental support that each arbitrary 13mer hybridizes with 7 bases to provide specificity, which represents 1/16,000 possible sequences, a theoretical calculation for the number of arbitrary primers needed to cover most of the expressed genes in a cell can be derived (Liang and Pardee, 1994, Methods Mol. Gen. 5:3). A given mRNA 3' end sequence of 600 bp resolved on a 6% denaturing polyacrylamide gel contains about 600 sites for recognition by all possible 16,000 7mers. Thus each arbitrary primer will have a 600 /16,000 = 4% probability of detecting any given mRNA. This permits an estimation of the number of arbitrary 13mers needed to be rationally designed in order to cover most of 10,000 expressed mRNA species in a mammalian cell. Others have claimed that fewer primers may cover most of the genes expressed, but those calculations were seriously flawed.
The number of arbitrary 13mers needed in combinations with all threeone-base anchored primers to detect a given fraction of mRNA by differential display.
n |
P = 1 - (0.96)n |
|
Comparison of Differential Display with other methodologies for cloning differentially expressed genes
Within 2 1/2 years of its invention in 1992, differential display (DD) quickly overtook subtractive hybridization to become the method of choice for cloning differentially expressed genes. Since then, despite competition from newer technologies such as arrays and SAGE, DD has continued to be the most commonly used technology for differential gene expression based on the number of publications that have been generated by each technology. GenHunter is proud to be the driving force for making differential display accessible to the biomedical research community.
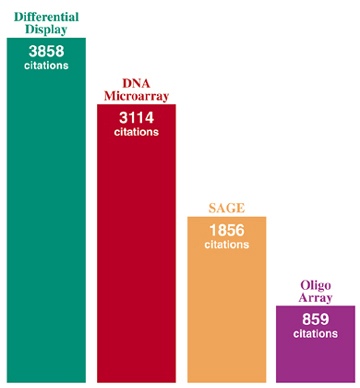 |
Publications using different methodologies for cloning differentially expressed genes. The number of Citations is the number of times the original publication has been cited by other papers, which reflects the number of times each technique has been used for publications. |
| Method | Year invented | Total | Pubs / Year | Original Reference |
| Differential Display | 1992 | 3858 | 257 | Science 1992, 257:967-971 |
| DNA Microarray | 1995 | 3114 | 259 | Science 1995, 270:467-470 |
| SAGE | 1995 | 1856 | 155 | Science 1995, 270:484-487 |
| Oligo Array | 1996 | 859 | 78 | Science 1996, 274:610-614 |
Search done with ISI Web of Knowledge Citation Search on January 4, 2007.
The advantages of DD over other technologies
Because of several major advantages, Differential Display (DD) technology continues to lead over other competing technologies such as DNA microarrays and SAGE in citation index and Medline hits (see figure above).
The advantages of DD include:
1) Simplicity: Technically, it is based on PCR and DNA sequencing gel electrophoresis.
2) Sensitivity: Five to ten micrograms of total RNA is enough to cover the majority of mRNAs in a eukaryotic cell. Also, DD is sequence-dependent. Therefore, if the sequences of the arbitrary primer match the target mRNA, whether abundant or rare, it will be amplified and detected. Other methodologies are mostly prevalence-dependent. Therefore, abundant messages are preferentially detected.
3) Reproducibility: Up to 99% of the bands on a mRNA display are reproducible. The accuracy can be even higher utilizing our Automated FDD Services.
4) Versatility: More than two RNA samples can be compared simultaneously allowing complex comparisons in the same experiment. Also, both up- and down-regulated genes can be detected at the same time. In addition, because the primers are randomly generated, the same kits can be used for any eukaryotic species.
5) Detecting Novel Genes: Unlike microarray, DD does not require prior knowledge of the mRNA sequence to be detected. Therefore, DD is considered an "open system" versus the "closed system" of Microarrays.
6) Speed and Expense: Within two days, the pattern of mRNA display can be obtained. Reamplification of probes and Northern blot confirmation take another week. Most importantly, since the assay may be checked at each step, it is no longer necessary, as it is with other methods, to wait until the end of the procedure to determine if it worked. Compared to DD, Microarray is not only expensive, but technically challenging for most labs.
Fundamental Differences between DD and DNA Microarrays
Given the great publicity of DNA microarrays techniques in recent years, many researchers have put their blind trust into this methodology, only to be overwhelmed by the confusing data generated. Knowing the major differences between differential display and DNA microarrays, both in theory and in practice, may help you to find the genes of real interest and save your valuable resources and effort.
Both differential display (DD) and microarrays are conceptually simple, however, the two methods are principally different. The fundamental difference is that differential display visualizes the mRNAs in subsets directly without any data normalization after their amplification and labeling by either isotopes or fluorescent dyes. In contrast, DNA microarrays visualize the mRNAs indirectly after the hybridization of extremely complex mRNA probes as first-strand cDNAs with fluorescent labels to a set of cDNA templates spotted on a glass surface. In fact, a cDNA probe used for microarray can be so complex that it consists of as many as 10,000 different species ranging from only a few copies to thousands of copies per cell. Further compounding the problem in signal specificity has been the fact that eukaryotic genes often come in families with many conserved sequences among the family members.
Therefore, lack of sensitivity, nonspecific- and cross-hybridization are major problems for microarrays. Also with microarrays, one is going to be limited to the detection of whatever genes that are spotted on a slide, making it a "closed’ system for gene discovery. In contrast, differential display is capable of detecting both known and novel genes with much higher gene coverage. Some of the most comprehensive commercial DNA microarrays, which contain a collection of approximately 10,000 human cDNAs, realistically only cover about 25% of the genome with 40,000 genes predicted. In fact the actual number of genes encoded by our genome could be much higher, since current computer programming is far from accurate in predicting genes from large genomes.
No matter which method is to be used, proper controls are absolutely critical before meaningful expression data can be obtained and analyzed. For example, cDNA pattern reproducibility is the key to reducing the rate of false positives for differential display. Likewise, labeling of each RNA sample being compared in microarrays with both red and green fluorescent dyes followed by reciprocal mixing them in color of fluorescence (e.g. RNA1-red/RNA2-green and RNA1-green/RNA2-red) before hybridization to duplicate slides is absolutely essential not only for data scoring, but also for data proof-reading. Although these important controls seem to be obvious for anyone serious about the quality of the microarray data, few published works actually have emphasized such controls.
One of the promising potentials of microarray is its capability of computerized data quantification and analysis. However, without knowing the actual quality of the data obtained, one could easily waste a lot of "clear" thinking on "dirty" data. The accuracy in gene expression analysis with microarrays is at best 3% or higher (3 out of 100 will be false positives), whereas GenHunter’s automated FDD service is at least 10 time more accurate.
Such confusing massive data generated by microarray has drawn sharp criticisms, which can be seen in our list of References listing problems with Microarrays (contact us to receive it).
Therefore, the choice is clear of whether you want to go to a "mine field" with hundreds of unconfirmed differences, or if you would like to find the few real "needles in the haystack".
Some of the major differences between differential display and microarrays are summarized in the following chart.
Comparison of Differential Display and DNA Microarrays
| Differential Display | DNA Microarray | |
| Year Invented | 1992 | 1995 |
| No. of Publications* | 3858 | 3114 |
| Publications/year | 257 | 259 |
| Detect novel genes | Yes | No |
| Compare more than 2 RNA samples | Yes | Mostly not |
| Signal detection | Directly (gel display) | Indirectly (by hybridization) |
| Total RNA required | 5 µg / 240 PCR reactions | >20 µg / hybridization |
| Gene coverage# | ~96% with 240 PCR reactions | ~25% with a 10K gene "chip" |
| Sensitivity (Detecting rare mRNA) | High (PCR based) | Low-medium |
| Immediate knowledge of gene sequence | No | Yes |
| Cost | Low-medium | High |
| Do it yourself? | Yes | Mostly not |
| Commercial services | GenHunter | Affymetrix, GE, Illumia, etc. |
* Publication search done with ISI Web of Knowledge Citation Search on January 4, 2007.
# Methods in Enzymology, 254:304-320, 1995.
For more information about our DD Kits and FDD Services, please click here.
In addition, a list of more than 500 selected DD publications, organized chronologically and by fields of application, shows the wide range of organisms and experiements that can be performed.GenHunter has also compiled a complete listing of references pointing out problems with Microarrays.
False positives in Differential Display
One of the most frequently asked questions for those who want to use differential display is what the false positive rate is. There is really no clear answer to this very complex question, because it depends on so many factors, both intrinsic and extrinsic, of the differential display method. The intrinsic factors include the quality of reagents and enzymes, type and purity of primers, integrity, concentration and purity of RNA used, etc. The extrinsic factors such as the systems being compared, experimental designs, appropriate internal controls, criteria for picking bands, reaction setup, type of PCR reaction tubes, type of thermocyclers, and, of course, the training and experience of a researcher, all can greatly contribute to the rate of false positives.
Taking an extrinsic factor, the system being compared, as an example, if one is to compare rat liver versus rat brain where it is known that nearly 50% of the genes expressed are brain specific, the false positives are going to be very low since the difference between samples compared are very large. However, if one is to compare gene expression between the same type of cells with and without a 30 min. treatment of a drug or hormone in hopes of identifying the immediate early genes, the false positives are going to be much higher simply due to the fewer differences in gene expression. Differential display itself can be perfectly reproducible if there are no intrinsic and extrinsic problems. For example, if you set up a 100 mL PCR reaction that is thoroughly mixed and aliquot 20 µL into each of 5 thin-walled PCR reaction tubes, the pattern of cDNA displayed among the five tubes can be perfectly the same. But if instead, 5 individual reactions are set up independently, the cDNA pattern amplified may not be the same simply due to pipetting errors. However, in practice, there is always going to be background "noise" due to pipetting errors because you have to compare different samples. These few spurious bands or "noise" can be as low as 0.1-1% of the cDNA bands displayed, which can be translated to 10-100 bands if you perform all 240 PCR reactions consisting of all the primer combinations in GenHunter's RNAimage® kits. As you can see, given the system that you will be comparing, the false positive rate can be estimated simply as the signal (truly differentially expressed genes) to noise (spurious, non-reproducible bands) ratio. Unlike the comparison between liver and brain, even if you encounter much higher false positives but are able to find the few immediate early genes induced by your drug or hormone, it would be a great accomplishment.
The other often-ignored extrinsic factor is the appropriate internal controls. Assuming that you are looking for the immediate early genes induced by a drug or hormone, you must make sure that the treatment truly worked based on either the phenotype of the cells, or more importantly, the induction of a known gene as a control. If this is not done, the differential display comparison is going to be flawed, because there may simply be no difference to begin with if the treatment did not work. In this case, if any differences are found, they are bound to be false positives. For a more detailed review on factors contributing to false positives in differential display, please see Liang and Pardee, 1994. Methods in Mol. Gen. 5:3.; Liang and Pardee, ed., 1997. Differential Display Methods and Protocols, Humana Press (Cat. No.: HP1).
There is no doubt that Differential Display is a very dependable method given its enormous popularity and the success of hundreds of thousands of researchers, thanks in part to GenHunter's complete line of reagent systems and step-by-step protocols (see representative publications in the back). Although you can count on GenHunter to solve the intrinsic problems of differential display, having a realistic understanding of the extrinsic problem areas will help you to minimize false positives and eliminate the fears and myths of differential display, one of the most powerful molecular biological tools ever invented.
Frequently used conversion factors
| Nucleic Acids | ||
| RNA | ||
| DNA (single-stranded) | ||
| DNA (double-stranded) |
Vendors for commonly used supplies not provided by GenHunter
| Taq DNA polymerase | ||
| Isotopes | ||
| Qiaex DNA extraction kit | ||
| Sequencing gel and buffers | ||
| Fluorescent Imager (FMBIO® II) |
